国家出手!口罩出口新规!附CE/FDA认证资格认证指南
来源: 时间:2020-04-02
商务部:4月1日起!出口医疗物资出新规


商务部昨晚发布公告:在疫情防控特殊时期,为有效支持全球抗击疫情,保证产品质量安全、规范出口秩序,自4月1日起,出口新型冠状病毒检测试剂、医用口罩、医用防护服、呼吸机、红外体温计的企业向海关报关时,须提供书面或电子声明(模版见附件1),承诺出口产品已取得我国医疗器械产品注册证书(相关注册信息见附件2),符合进口国(地区)的质量标准要求。海关凭药品监督管理部门批准的医疗器械产品注册证书验放。上述医疗物资出口质量监管措施将视疫情发展情况动态调整。
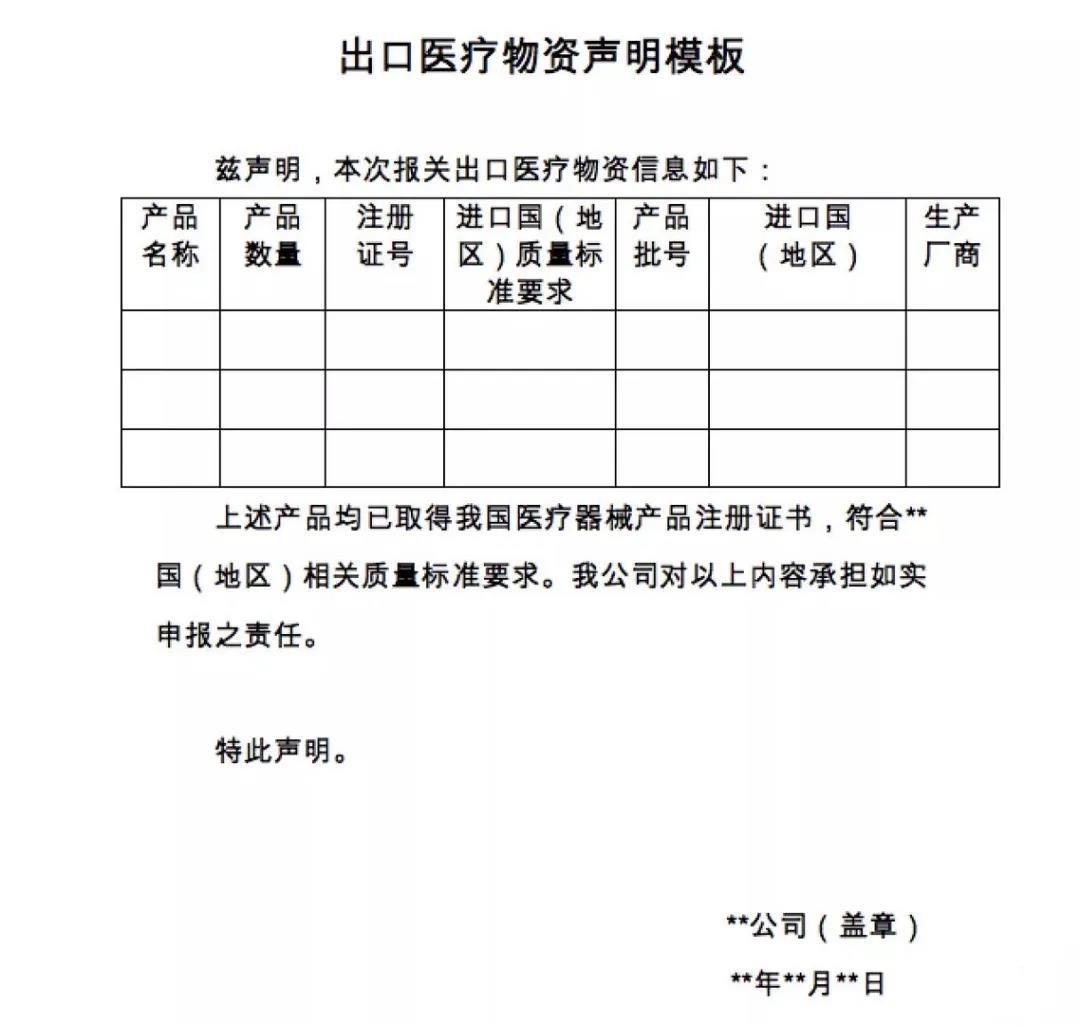
▲出口报关时新增的申明模板
根据国家商务部,海关总署和国家药品监督管理局官方公布的已经取得我国医疗器械产品注册证书的合格企业,全国共计2047家!
大家在询价,采购和出口时,可以仔细从中查找你的供应商是否在三个部委公布的合规清单里面!
避免采购劣质产品,导致货物被查被扣并最终血本无归!
一次性使用医用口罩(共752家)
医用防护口罩(共150家)
医用外科口罩(共523家)
医用防护服(共301家)
医用防护口罩(共23家)
呼吸机(共62家)
红外体温计(共236家)
欧盟CE认证资格
随着新冠肺炎疫情的迅速蔓延,意大利、法国、德国等国纷纷沦陷,欧洲成为了疫情重灾区。在防控疫情的过程中,由于医用口罩、防护服等医护物资的严重短缺,欧洲对外释放了巨大的防护用品需求。目前,中国已经取得了新冠防疫的阶段性胜利,作为世界上最大的医用防护用品生产国,无论是传统医疗生产企业,还是决定踏入这一陌生领域的新企业,都决定将多余的产能利用起来,将此类产品销往欧洲市场。
出口欧盟市场,CE认证必不可少。目前市场上出现了各色各样的医疗CE证书,让人眼花缭乱。在各种行业微信群里,经常可以看到有人发出一张所谓的CE证书,请大家帮忙辨别真伪。为了更好地帮助到大家,下面我们就来谈谈具体的鉴别方法。
查询CE证书的真伪,有多种方式,首先是最简单粗暴的一种。大的公告机构会在自己的官网上开放查询证书的窗口,当然,这种方式仅适用于发证机构正好提供了查询服务的情况。而对于未开放证书查询服务的机构,就不会奏效了。那么对于此类情况,当我们拿到一张医疗CE证书时,我们又该如何鉴别呢?
欧盟官网MDD 93/42/EEC医疗器械指令授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13
欧盟官网MDR (EU) 2017/745医疗器械法规授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
欧盟官网 (EU)2016/425个人防护装备授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=15550

自2020年5月26日起,MDR (EU) 2017/745医疗器械法规将正式取代欧盟现行的MDD医疗器械指令强制实施,同样在欧盟官网可以查询到,拥有MDR授权的公告机构目前只有12家。
所以,如果您手上的医疗CE证书发证机构不在以上名单范围内,则说明它并不具备医疗产品的欧盟发证资质,更别谈CE证书的发放了,那么,很遗憾地说,您拿到的这张“”CE证书“是无效的。
另外,我们也可以从医疗器械产品CE认证的流程着手去分析,完成鉴别。

美国FDA
510(k)的申请流程如下:

通常一个产品从启动510(k),准备测试和各类文件,直至最终的审核结束,需要历经8-10个月,而初次申请的企业,这个时间通常会更长。
除了510(k)以外,FDA要求所有的医疗器械企业都需要进行场所注册(Establishment Registration)和产品列示(Product Listing),这一要求对于应急使用授权的产品也不例外。对于所有的海外企业,在进行场所注册之前应先获得邓白氏编码,编码在中国由华夏邓白氏公司代理发放,免费的代码需要大约30天可以获得。获取邓白氏编码后,大约需要1-2周左右完成场所注册和产品列示。
无论是510(k),场所注册或者产品列示,FDA都不会向企业颁发任何证书,仅以美国FDA数据库中的数据为准。也就是说,大家见过的各种有着老鹰标记的证书都是没有任何效力的。
目前,新冠疫情在美国呈现明显爆发的趋势,各类医疗物资也趋于紧张。美国食品药品监督管理局(FDA)早在今年(2020年)2月初就开始对医疗器械潜在短缺的情况进行调查,为了应对各类医疗器械的紧缺FDA发布了各类应急使用授权(Emergency Use Authorization,EUA)。目前可以申请EUA的产品主要是未获上市的N95口罩、未获上市的新冠病毒诊断试剂、未获上市的酒精洗手液产品、已上市但需扩展用途的非侵入远程监护系统、已上市和未上市的呼吸设备。
新冠检测试剂:首当其冲的就是诊断试剂,FDA已经就该产品发布了第二版的指导原则。第一版指导原则主要针对美国国内临床实验室自我开发的检验方法的EUA申请,而第二版则纳入了针对生厂商的递交EUA申请的详细指导。EUA的申请要包含的内容与510(k)相似,需要提交检测试剂的描述、预期用途、性能评价报告、临床评价方案、稳定性试验方案、标签标识等。由于属于应急审批,FDA还要求企业必须提供针对患者和专业人员的明白纸(Fact Sheet)。目前海河咨询已经完成了新冠病毒胶检测试剂盒胶体金法的EUA申请。
(2)经NIOSH批准但已过制造商推荐的保存期限的过滤面罩口罩,供医护人员在医疗环境中使用,以防止医护人员由于面罩口罩短缺,而暴露于病原性生物空气传播颗粒中。
考虑到这样的措施还是无法保障美国市场的口罩供应,FDA又在近期发布口罩的EUA申请,特别地,没有获得NIOSH批准的口罩也可以进行EUA的申请,但是必须符合:
条件1-满足特定国家/地区性能标准,包括
澳大利亚:P2,P3
巴西:PFF2,PFF3
欧盟:FFP2,FFP3
日本:DS/DL3,DS/DL2
韩国:Special 1st
墨西哥:N100, P100, R100, N99, P99, R99, N95, P95, R95
条件2-获得特定国家上市许可,包括:
欧盟:CE认证;
澳大利亚:ARTG注册
加拿大:注册证;
日本:医疗器械注册证。
酒精洗手液产品:之前酒精洗手液产品在美国是按照非处方类药品进行管理的,洗手液中的酒精则是按照API进行管理的。目前,FDA已经发布通知,不会对提供用于洗手液的酒精的生产商或者洗手液的生产商采取行动,即可以认为,FDA将允许用于洗手液的酒精和基于酒精的洗手液在满足限定条件下直接在美国销售。海河咨询特别提醒:对于洗手液的生产商仅限定适用于药剂师以及联邦设施,不适用于其他商业生产商。
★ 口罩出口--海关技术性贸易措施指南 ★
一、出口通关提示
1.报关前提条件
收发货人注册编码(慈善机构可为临时编码),需办理无纸化通关法人卡
2.出口资质
口罩出口对生产销售单位、境内发货人,除满足国内生产、市场流通资质需求外,中国海关无特殊资质要求。
3.出口申报要求
(1)商品归类:除特殊情况外,绝大部分口罩应归入税号63079000。
(2)检验检疫:口罩为非法检产品,申报时检验检疫项目无需填报。根据我国政府与相关国家签订的政府间检验协议,对出口伊朗等少数几个国家的产品需按规定进行装运前检验。
(3)关税征免:如出口物资为贸易性质,征免性质申报一般征税,征免方式申报照章征税;如为捐赠性质,境内发货人为贸易代理商、慈善机构等,征免性质可不填,征免方式申报全免。
(4)禁限管理:目前商务部未对口罩设置贸易管制要求,中国海关也无针对防护物资的监管证件口岸验核要求。
(5)申报规范:按照规范申报要求填写商品名称、成分含量;如物资非中国生产,原产国按照实际生产国填写。
4.出口退税
口罩的出口退税率为13%。
5.中美关税排除加征
美国企业可申请排除口罩进口加征关税,但是目前只有少数企业获准豁免。详见美国贸易代表办公室网站https://ustr.gov/。
6.快速通关保障
物资出口申报如遇单窗等系统故障,可联系现场海关采取应急方式处置,或者拨打海关12360热线进行咨询。
*以下内容是根据国内外相关政府机构、专业网站、新闻报道,收集整理而成,仅供参考。具体内容以相关管理部门、国外官方机构要求为准。
二、出口前准备
1.明确口罩分类
国外按照用途一般分为个人防护和医用两类口罩,国内出口贸易企业需具备的资质和材料如下:
(1)营业执照(经营范围有相关经营内容)。
(2)企业生产许可证(生产企业)。
(3)产品检验报告(生产企业)。
(4)医疗器械注册证(非医用不需要)。
(5)产品说明书(跟着产品提供)、标签(随附产品提供)。
(6)产品批次/号(外包装)。
(7)产品质量安全书或合格证(跟着产品提供)。
(8)产品样品图片及外包装图片。
(9)贸易公司须取得海关收发货人注册备案。
2.国内出口口罩生产企业资质证明
生产个人防护或者工业用非医疗器械管理的普通口罩,有进出口权的企业,可自行直接出口。
生产属于医疗器械管理的口罩用于出口,中国海关不需要企业提供相关资质证明文件,但一般进口国会要求生产企业提供产品三证,以证明该进口的商品在中国已合法上市,具体如下:
(1)营业执照(经营范围包含有医疗器械相关,非医疗级别的物品不需要)。
(2)医疗器械产品备案证或者注册证。
(3)厂家检测报告。
生产企业有进出口权,可以自行出口,如没有进出口权,可以通过外贸代理进行出口销售。
3.内贸企业做出口需要取得的基本资质
(1)向市场监管部门取得营业执照,增加经营范围“货物进出口、技术进出口、代理进出口”。
(2)向商务部门取得进出口权,可直接在商务部业务系统统一平台(http://iecms.mofcom.gov.cn/)申请,网上提交材料。
(3)向外汇管理局申请取得开设外汇账户许可。
(4)办理进出口货物收发货人海关注册登记。
三、各国口罩准入条件(产品准入条件)
1.美国
必要资料:提单,箱单,发票。
个人防护口罩:必须取得美国 NIOSH检测认证,即National Institute for Occupational Safety and Health美国国家职业安全卫生研究所认证。
医用口罩:须取得美国FDA注册许可。
2.欧盟
必要资料:提单,箱单,发票。
个人防护口罩:个人防护口罩的欧盟标准是EN149,按照标准将口罩分为FFP1/FFP2和FFP3三个类别。所有出口欧盟的口罩必须获得CE认证证书。CE认证是欧盟实行的强制性产品安全认证制度,目的是为了保障欧盟国家人民的生命财产安全。
医用口罩:医用口罩对应的欧盟标准是EN14683。
产品在欧盟销售需要出具欧盟自由销售证书 Free Sale Certificate,有了CE标志并进行了相关指令中要求的欧盟注册后,中国的制造商出口欧盟不需要自由销售证书。
3.日本
必要资料:提单,箱单,发票,日本国外的制造商必须向PMDA注册制造商信息。
口罩包装要求:包装上印有ウィルスカット(中文翻译:病毒拦截)99%的字样
PFE:0.1um微粒子颗粒过滤效率
BFE:细菌过滤率
VFE:病毒过滤率
口罩品质标准:
(1)医用防护口罩:符合中国GB 19083-2010 强制性标准,过滤效率≥95%(使用非油性颗粒物测试)。
(2) N95口罩:美国NIOSH认证,非油性颗粒物过滤效率≥95%。
(3) KN95口罩:符合中国GB 2626 强制性标准,非油性颗粒物过滤效率≥95%。
4.韩国
必要资料:提单,箱单,发票,韩国进口商营业执照。
个人防护口罩标准:KF (Korean filter) 系列分为KF80、KF94、KF99
执行标准规范:MFDS Notice No. 2015-69
韩国医疗器械准入的法规门槛,基本分类为I、II、III、IV类,持证为韩国公司(License holder),韩国收货人需要到韩国药监局Korea Pharmaceutical Traders Association. 提前备案进口资质(没有不行)网址:www.kpta.or.kr。
5.澳大利亚
必要资料:提单,箱单,发票。
须通过澳洲的TGA注册,符合标准规范:AS/NZS 1716:2012,此规范是澳大利亚和新西兰的呼吸保护装置标准。
TGA 是Therapeutic Goods Administration的简写,全称是治疗商品管理局,它是澳大利亚的治疗商品(包括药物、医疗器械、基因科技和血液制品)的监督机构。澳大利亚对医疗器械分为I类,Is and Im, IIa, IIb, III类,产品的分类几乎和欧盟分类一致,如果产品已经获得CE标志,则产品类别可以按照CE分类。
四、各国注册、认证简要办理流程
1.美国NIOSH认证
需按照NIOSH的指南实施,企业需寄送样品至NIOSH实验室实施测试,同时提交技术性资料(包括质量体系部分资料)至NIOSH文审,只有文审和测试都通过,NIOSH才核发批文。NIOSH将其认证的防颗粒物口罩分为9类,具体的测试则由NIOSH下属的NPPTL (National Personal Protective Technology Laboratory)实验室操作。主要测试指标包括呼气阻力测试、呼气阀泄漏测试、吸气阻力测试、过滤效率测试。
2.美国FDA注册

3.欧盟CE注册

4.日本PMDA注册

(1)准备阶段。确定产品分类(I,II特殊控制,II类控制,III,IV)和产品JMDN编码,选择MAH(日本持证方);
(2)制造商向PMDA注册工厂;
(3)II类特殊控制产品向授权认证机构PCB申请QMS工厂审核,其他II类产品和III类IV类产品向PMDA申请QMS工厂审核,并获得QMS证书;
(4)申请Pre-Market Apporval证书,II类特殊控制由PCB发证,其他II类产品和III类IV类产品控制由MHLW(厚生劳动省)发证;
(5) 支付申请费用;
(6) 注册文件整改,注册批准;
(7)所有类别产品均需要MAH向RBHW(厚生省地区机构)进行进口通报注册后才能进口销售。
5.韩国KFDA注册
韩国卫生福利部(MinistryofHealthandWelfare,MHW),简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是最主要的卫生保健部门。依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部负责对医疗器械的监管工作。KFDA注册流程为:
(1)确定产品分类(I,II,III,IV),选择KLH(韩国持证方);
(2)II类产品需申请KGMP证书和接受现场审核,II类产品一般是授权的第三方审核员,并获得KGMP证书;
(3)II类产品需要送样品到韩国MFDS授权的实验室进行韩国标准的测试;
(4)由KLH向MFDS(韩国食品药品安全部)提交技术文件(检测报告,KGMP证书等),进行注册审批;
(5)支付申请费用;
(6)注册文件整改,注册批准;
(7)指定韩国代理商和经销商,产品销售。
6.澳大利亚TGA注册
依据Australian Therapeutic Goods (Medical Devices) Regulations 2002,澳大利亚对医疗器械分为I类,Is and Im, IIa, IIb, III类,产品的分类几乎和欧盟分类一致,如果产品已经获得CE标志,则产品类别可以按照CE分类。如果已经获得欧盟公告机构(Notified Body)签发的CE证书,是可以被TGA认可的,并可以作为满足澳大利亚安全法规的重要注册资料。

五、各国口罩技术标准对比(供生产企业参考)



六、各国口罩技术标准(供生产企业参考)
|
序号 |
标准号 |
标准名称 |
状态 |
发布时间 |
|
国际 |
ISO 22609:2004 |
传染试剂防护服.医疗面罩.防人造血渗透的试验方法(固定容积,水平注射) |
现行 |
2004/12/3 |
|
欧盟 |
EN 136-1998 |
呼吸保护装置.全面罩.要求,试验,标记。 |
现行 |
1998/1/1 |
|
EN 140-1998+AC-1999 |
呼吸保护装置.半面罩和四分之一面罩.要求,试验和标记。 |
现行 |
1998/9/1 |
|
|
EN 143-2000 |
呼吸防护装置.微粒过滤器.要求,试验,标记。 |
现行 |
2000/2/1 |
|
|
EN 149-2001 |
呼吸防护装置.颗粒防护用过滤半面罩.要求,检验和标记。 |
现行 |
2001/4/1 |
|
|
EN 529-2005 |
呼吸保护装置.选择,使用,保养和维修的建议。 |
现行 |
2005 |
|
|
EN 12942-1998 |
呼吸保护器.带全面罩,半面罩和四分之一面罩的鼓风过滤装置.要求,检验,标识。 |
现行 |
1998 |
|
|
EN 14387-2004+A1-2008 |
呼吸保护装置.气体过滤器和组合过滤器.要求、测试、标记。 |
现行 |
2004/1/1 |
|
|
EN 14683-2019 |
医用口罩 要求和试验方法。 |
现行 |
2019/3/1 |
|
|
美国 |
ASTM F1862/F1862M-2017 |
医用口罩抗人工合成血渗透的标准试验方法(已知速度下固定体积的水平投影)。 |
现行 |
2017 |
|
ASTM F2100-2019 |
医用口罩材料性能标准规范。 |
现行 |
2019 |
|
|
ASTM F2101-2019 |
用金黄色葡萄球菌生物气溶胶评价医用口罩材料的细菌过滤效率(BFE)的标准试验方法。 |
现行 |
2019 |
|
|
ASTM F2299/F2299M-2003(2017) |
用胶乳球测定医用面具材料粒子渗透性初始效率的标准试验方法。 |
现行 |
2003 |
|
|
澳大利亚 |
AS/NZS 1715:2009 |
呼吸保护设备的选择,使用和维护。 |
现行 |
2009/2/6 |
|
AS/NZS 1716:2012 |
呼吸保护装置。 |
现行 |
2012/2/13 |
|
|
日本 |
JIS T 8062:2010 |
预防传染性病原体的防护服.面罩.防止人造血浆渗透的试验方法(确定容量,平行注射)。 |
现行 |
2010/5/25 |
|
JIS T 8159:2006 |
呼吸防护设备的选择、使用和维护指南。 |
现行 |
2006/4/25 |
|
|
JIS T 8159:2006 |
呼吸保护装置泄漏率试验方法。 |
现行 |
2006/2/20 |
|
|
韩国 |
KS M 6673-2008 |
防尘口罩 |
现行 |
2008/2/22 |
|
KS K ISO 22609-2018 |
传染试剂防护服.医疗面罩.防人造血渗透的试验方法(固定容积、水平喷射)。 |
现行 |
2018/11/14 |
*以上技术标准如有动态调整,以相关标准管理机构官方发布为准。
Copyright @ 2017 山东省信达雅国际商贸有限公司版权所有 . 鲁ICP备11006413号-1
电话: 0531-86080340 | 86086106 | 86086107
地址:济南市泉城路院前街珍珠泉大院(省人大大院)9号楼5楼













